Description of SGRF
SGRF is a model for variant-aware gene structure prediction. It is distributed as part of the ACE
package, which interprets genetic variants that alter gene structure (either by
modifying translation reading frames or by modifying splicing
patterns), in terms of their effect on encoded proteins or potential to
cause a loss of function.
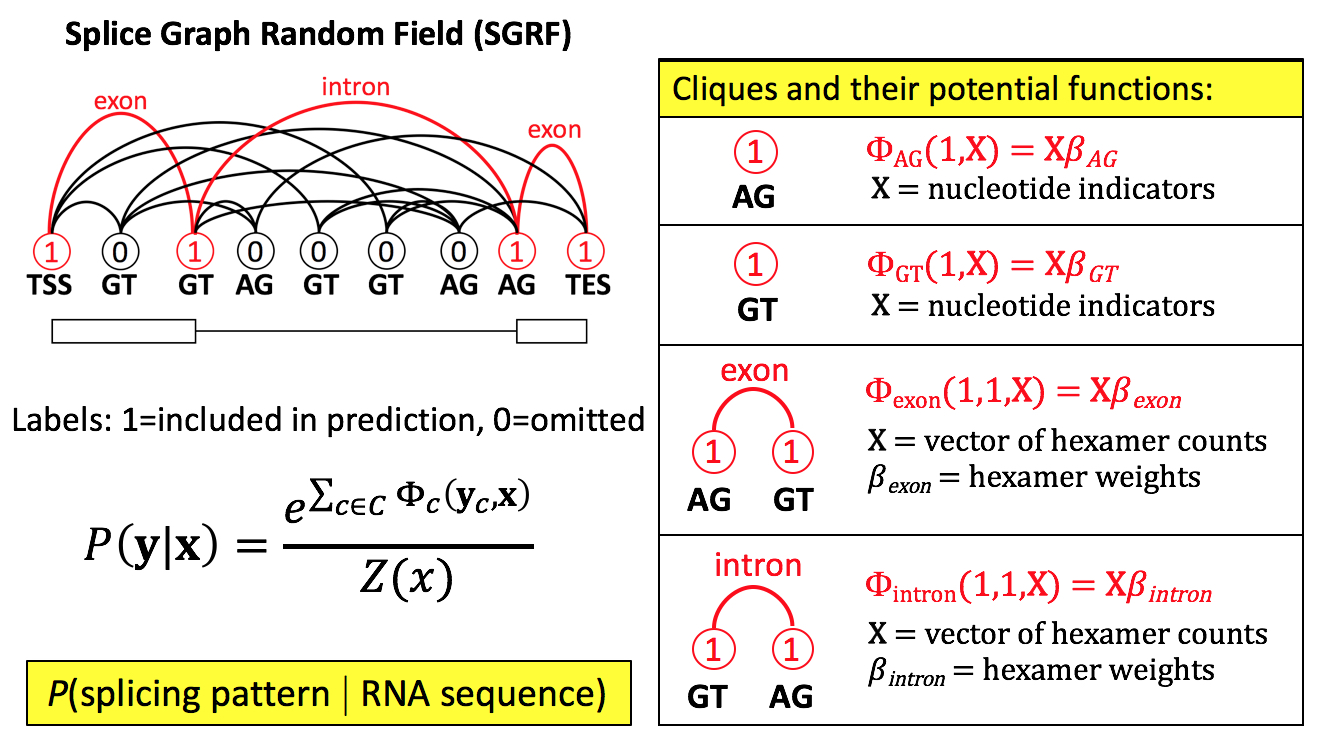
SGRF stands for splice graph random field, a type of probabilistic grammar model. The SGRF assigns a probability to each predicted splicing pattern, allowing putative splicing patterns to be ranked by their probability of being used by the cell. The ACE package can then be used to interpret, for each isoform, whether that isoform is likely to result in loss or change of function.
SGRF stands for splice graph random field, a type of probabilistic grammar model. The SGRF assigns a probability to each predicted splicing pattern, allowing putative splicing patterns to be ranked by their probability of being used by the cell. The ACE package can then be used to interpret, for each isoform, whether that isoform is likely to result in loss or change of function.
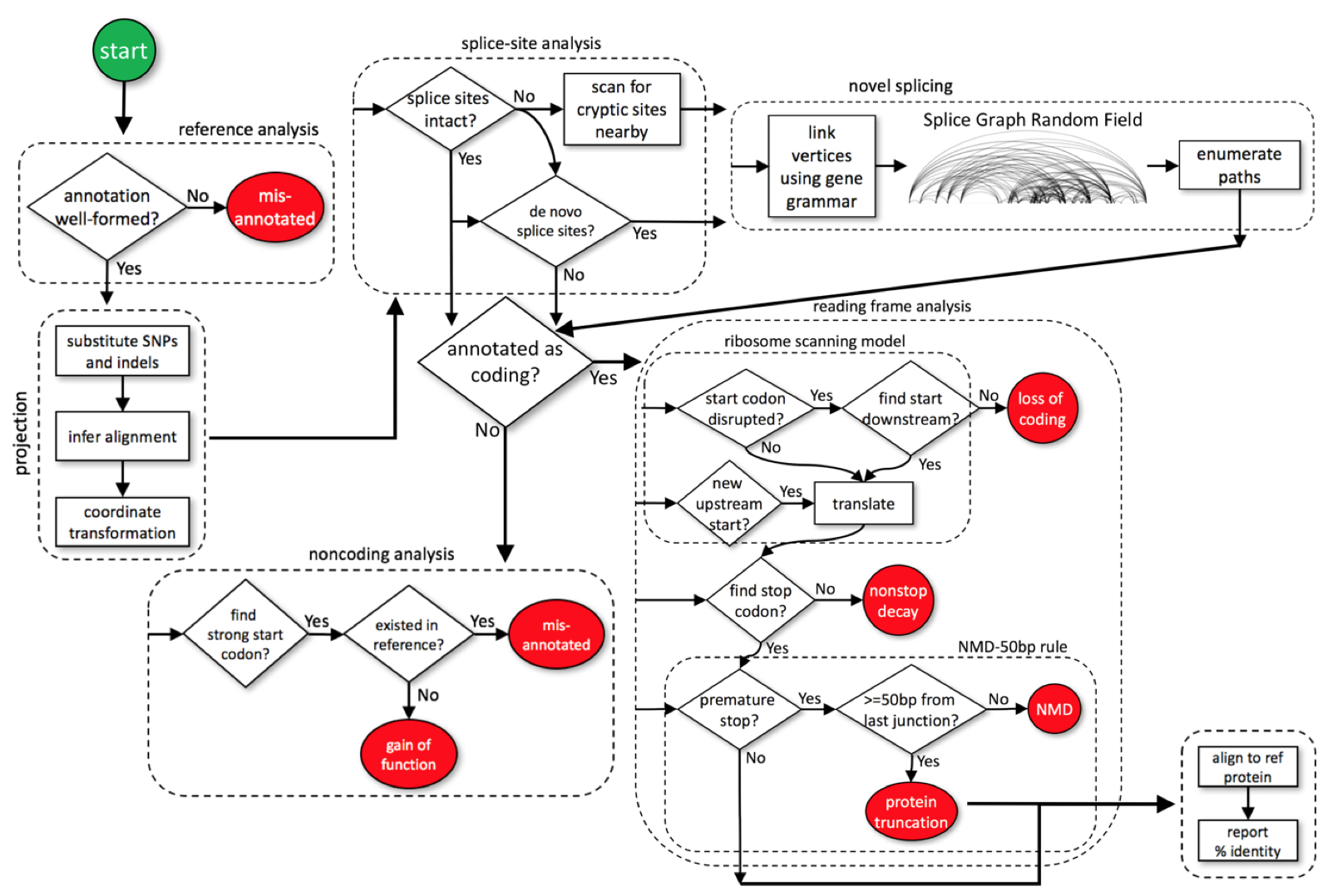
Unlike traditional
gene finders that key primarily
on coding signals, SGRF instead models the process of exon definition.
Exon definition features, including splicing enhancers and splicing
silencers, modulate the effective strengths of individual splice
sites. The cell thus makes splicing decisions not based solely on
splice sites, but based on the aggregate influence of splice sites and
the splicing enhancers and silencers within exons and introns flanking
those splice sites. SGRF models the joint effect of splice sites
and
splicing regulatory elements. Because it does not
rely on coding signals, SGRF can predict splicing changes that
interrupt translation reading frames.
In order to apply SGRF to non-human species, it must first be trained. The three parts of SGRF that need to be trained are (1) the donor splice site model, (2) the acceptor splice site model, and (3) the exon definition model. These training steps are described below.
The following must be installed prior to using SGRF:
exons.model : logistic regression exon model
introns.model : logistic regression intron model
logreg-donors.model : logistic regression donor site model
logreg-acceptors.model : logistic regression acceptor site model
intergenic.model : intergenic model
Please ensure that these models are properly referenced in the configuration file. See below for instructions on editing the configuration file.
For non-human data, you need to train models specific to your target organism. To train the splice-site models, you first need to extract training examples from a set of gene annotations. Given a FASTA file genome.fasta containing entries for each chromosome, and a GTF (GFF version 2, not version 3!) file anno.gtf containing annotated exons and introns, the following command will extract training examples for donor and acceptor splice sites:
Note that the substrate field (the first field) in the GTF file must match the identifier after the ">" in the FASTA file. The parameter GT,GC,AT is a list of the allowable consensus sequences for donor splice sites, and AG,AC is a list of allowable consensus sequences for acceptor splice sites. Note that there can be no spaces within those consensus lists. The ATG and TGA,TAA,TAG parameters give start and stop codon lists.
The above command will extract only positive examples. The following script will extract negative examples. This script takes no parameters, because it extracts the examples from files generated by the previous command:
After executing the above commands, the files donors.fasta and acceptors.fasta will contain the positive examples, and the files non-donors.fasta and non-acceptors.fasta will contain the negative examples.
These files should contain a series of 162 bp sequences, each one with a splice site in the center:
>ENST00000342066
AGACGCTTATGTCCAAGGGGATCCTGCAGGTGCATCCTCCGATCTGCGACTGCCCGGGCT
GCCGAATATCCTCCCCGGTGGTGAGATGCGGGGCTCGGTTGGGGCTGGGAGTTACTCTCC
CCTGCGGAGCTTGTCCCTGCGGTTTTCAGGGTTTTCAGGATC
...etc...
Now you can train each classifier (one for donor splice sites and one for acceptor splice sites) using the logreg-splice.py program, which has the following usage:
The left-context parameter is an integer specifying the number of nucleotides to the left (5' of) the consensus sequence to include in the model. Simiarly, the right-context specifies how many nucleotides to include after the consensus. The alpha parameter controls regularization: 0 results in ridge regression, 1 results in lasso, and values between 0 and 1 result in elastic net regression. We recommend a default value of 0.5. The consensus list can be copied from the earlier command for get-examples.pl. The final parameter (type) should be GT for the donor splice site model and AG for the acceptor splice site model; this parameter is merely an indicator of the splice site type, not a specification of the consensus sequence.
For example, the following commands will train a donor splice site model and an acceptor splice site model:
Training the exon definition model consists of three steps: (1) extracting hexamer features from training examples; (2) using logistic regression to learn feature weights; (3) installing those feature weights into a model file.
The first step, extracting hexamer features from training examples, can be done using this command:
The files internal-exons.fasta and introns.fasta should have been created by the get-examples.pl script that was executed earlier (see above). After the command finishes, the file hex-counts.txt should contain a list of all hexamers followed by tab-separated fields giving hexamer counts (where many of the hexamer counts will be 0.0):
Now you can train the exon definition model by running the R script logistic-regression.R, which has the following usage:
For example:
$ACEPLUS/logistic-regression.R hex-counts.txt 0.5 betas.txt
NOTE: if this script fails to execute, you might not have Rscript installed, or your R environment might not be properly set up. In that case, you can run this script via the command "R CMD $ACEPLUS/logistic-regression.R hex-counts.txt 0.5 betas.txt". You may need to specify a full path to the R executable on your system.
Alpha specifies the type of regularization, just as with the logreg-splice.py command earlier; we recommend a default value of 0.5 to equally interpolate between ridge regression (0.0) and lasso regression (1.0). This script may run for many hours, depending on the number of training examples given. When running on 10,000 exons and 10,000 introns, we have found that the script takes approximately 12 hours on a single CPU core. When the script finishes, the learned hexamer weights should be in the betas.txt file:
4097 x 1 sparse Matrix of class "dgCMatrix"
1
(Intercept) 3.41155949
AAAAAA -47.12145843
AAAAAC .
AAAAAG 42.45011428
AAAAAT .
AAAACA -3.10915854
AAAACC .
AAAACG 0.86292402
...etc...
Now these weights need to be installed into model files for exons and introns. Because the intron weights are simply the negatives of the exon weights, the same betas.txt file generated above can be used for both exons and introns:
After verifying that the generated model files are non-empty, please copy them into the directory where your configuration script is located (see the next section).
For parameters that name a file, please give full absolute paths (unless you are using Docker, in which case you should omit paths, place all files in the current directory, and specify the vcf directory as /root).
min-path-score : Any gene prediction that scores less than this value will be omitted from the output of SGRF. The lowest possible score is 0.0, which is the default threshold. The highest possible value is 1.0.
max-alt-structures : This is the number of alternate splicing patterns that can be predicted by SGRF.
The remaining parameters are described on the ACE web page. Note that human model files for SGRF are included in the git distribution for ACE. If your target organism is human, you can use these files:
exons.model : logistic regression exon model
introns.model : logistic regression intron model
logreg-donors.model : logistic regression donor site model
logreg-acceptors.model : logistic regression acceptor site model
intergenic.model : intergenic model
If your target organism is not human, please retrain SGRF by following the instructions above on training the splice site models and the exon definition model. Note that the intergenic model need not be retrained, since SGRF cannot predict changes to the intergenic region.
The usage of this command is nearly identical to the usage of the original ACE command. Please see the original ACE web page for instructions on setting these parameters and interpreting the outputs.
Before running SGRF, you will need to make the personal genome (alt.fasta) using the make-individual-genomes.pl script, as described on the original ACE web page.
In order to apply SGRF to non-human species, it must first be trained. The three parts of SGRF that need to be trained are (1) the donor splice site model, (2) the acceptor splice site model, and (3) the exon definition model. These training steps are described below.
Prerequsites
|
UPDATE: If you encounter difficulty installing SGRF on your system, you can now install SGRF via Docker. |
The following must be installed prior to using SGRF:
- ACE.
Please follow the
instructions for installing ACE on the
ACE web
page.
Follow the instructions on that page for setting environment variables
and installing perl and python version 3.5 or higher. You will
need g++ to
compile ACE, and the Gnu Scientific Library (gsl).
- R. Please download and install R from www.r-project.org.
- glmnet (R
package). Please download and install glmnet from CRAN.
Installing via Docker
To install SGRF via a
Docker image, you first need to install
Docker on your system.
Once Docker is installed and the app is running, you can either
download the 3.7 GB pre-compiled image file, or build the image
yourself
from a Dockerfile. To download the pre-compiled image:
If you instead prefer to build a docker image yourself, download the
Once you have a compiled image (either downloaded via
where X is the absolute path to the current directory where you're working, and "..." is the command to be executed. For example, to train a donor splice site model, the Dockerized command would be:
where DIR is the absolute path to the current directory where you're working.
docker pull
genezilla/aceplusIf you instead prefer to build a docker image yourself, download the
Dockerfile
for SGRF and build it using the docker build command:wget
https://raw.githubusercontent.com\
/bmajoros/ACE/master/Dockerfile
mkdir aceplus
mv Dockerfile aceplus
docker build -t genezilla/aceplus aceplusOnce you have a compiled image (either downloaded via
docker pull
or built manually using docker build), you can execute
any SGRF or ACE command by first prefixing it with: docker run -w
/root -v X:/root -it
genezilla/aceplus ...where X is the absolute path to the current directory where you're working, and "..." is the command to be executed. For example, to train a donor splice site model, the Dockerized command would be:
docker
run -w /root -v DIR:/root -it genezilla/aceplus logreg-splice.py donors.fasta non-donors.fasta 6
12 0.5 GT,GC,AT GT > donors.modelwhere DIR is the absolute path to the current directory where you're working.
|
UPDATE: There is now a whole page on running SGRF via docker, with extensive examples of Docker commands. |
Training the Splice Site Models
To run SGRF on human data, you can use the existing models included in the ACE package:exons.model : logistic regression exon model
introns.model : logistic regression intron model
logreg-donors.model : logistic regression donor site model
logreg-acceptors.model : logistic regression acceptor site model
intergenic.model : intergenic model
Please ensure that these models are properly referenced in the configuration file. See below for instructions on editing the configuration file.
For non-human data, you need to train models specific to your target organism. To train the splice-site models, you first need to extract training examples from a set of gene annotations. Given a FASTA file genome.fasta containing entries for each chromosome, and a GTF (GFF version 2, not version 3!) file anno.gtf containing annotated exons and introns, the following command will extract training examples for donor and acceptor splice sites:
$ACEPLUS/get-examples.pl
anno.gtf genome.fasta GT,GC,AT AG,AC ATG
TGA,TAA,TAG
Note that the substrate field (the first field) in the GTF file must match the identifier after the ">" in the FASTA file. The parameter GT,GC,AT is a list of the allowable consensus sequences for donor splice sites, and AG,AC is a list of allowable consensus sequences for acceptor splice sites. Note that there can be no spaces within those consensus lists. The ATG and TGA,TAA,TAG parameters give start and stop codon lists.
The above command will extract only positive examples. The following script will extract negative examples. This script takes no parameters, because it extracts the examples from files generated by the previous command:
$ACEPLUS/get-negative-examples.pl
After executing the above commands, the files donors.fasta and acceptors.fasta will contain the positive examples, and the files non-donors.fasta and non-acceptors.fasta will contain the negative examples.
These files should contain a series of 162 bp sequences, each one with a splice site in the center:
>ENST00000342066
AGACGCTTATGTCCAAGGGGATCCTGCAGGTGCATCCTCCGATCTGCGACTGCCCGGGCT
GCCGAATATCCTCCCCGGTGGTGAGATGCGGGGCTCGGTTGGGGCTGGGAGTTACTCTCC
CCTGCGGAGCTTGTCCCTGCGGTTTTCAGGGTTTTCAGGATC
...etc...
Now you can train each classifier (one for donor splice sites and one for acceptor splice sites) using the logreg-splice.py program, which has the following usage:
$ACEPLUS/logreg-splice.py
<pos-fasta> <neg-fasta> <left-context>
<right-context>
<alpha> <consensus-list> <type>
The left-context parameter is an integer specifying the number of nucleotides to the left (5' of) the consensus sequence to include in the model. Simiarly, the right-context specifies how many nucleotides to include after the consensus. The alpha parameter controls regularization: 0 results in ridge regression, 1 results in lasso, and values between 0 and 1 result in elastic net regression. We recommend a default value of 0.5. The consensus list can be copied from the earlier command for get-examples.pl. The final parameter (type) should be GT for the donor splice site model and AG for the acceptor splice site model; this parameter is merely an indicator of the splice site type, not a specification of the consensus sequence.
For example, the following commands will train a donor splice site model and an acceptor splice site model:
$ACEPLUS/logreg-splice.py
donors.fasta non-donors.fasta 6 12 0.5 GT,GC,AT GT > donors.model
$ACEPLUS/logreg-splice.py acceptors.fasta non-acceptors.fasta 20 2 0.5 AG,AC AG > acceptors.model
Please copy the resulting model files into a model
directory where your configuration script is located; see the section
below on editing the default
configuration script.$ACEPLUS/logreg-splice.py acceptors.fasta non-acceptors.fasta 20 2 0.5 AG,AC AG > acceptors.model
Training the Exon Definition Model
To run SGRF on human data, you can use the existing models included in the ACE package, as noted above. To run SGRF on non-human data, you need to retrain the splice-site models (described above) and the exon/intron definition models, as described here.Training the exon definition model consists of three steps: (1) extracting hexamer features from training examples; (2) using logistic regression to learn feature weights; (3) installing those feature weights into a model file.
The first step, extracting hexamer features from training examples, can be done using this command:
$ACEPLUS/get-hexamer-counts.py
internal-exons.fasta introns.fasta > hex-counts.txt
The files internal-exons.fasta and introns.fasta should have been created by the get-examples.pl script that was executed earlier (see above). After the command finishes, the file hex-counts.txt should contain a list of all hexamers followed by tab-separated fields giving hexamer counts (where many of the hexamer counts will be 0.0):
category
AAAAAA AAAAAC AAAAAG
AAAAAT AAAACA AAAACC
AAAACG AAAACT AAAAGA
AAAAGC AAAAGG AAAAGT ...etc...
1 0.0 0.0 0.0 0.0 0.0 1.0 0.0 0.0 ...etc...
1 0.0 0.0 0.0 0.0 0.0 1.0 0.0 0.0 ...etc...
Now you can train the exon definition model by running the R script logistic-regression.R, which has the following usage:
logistic-regression.R
<hex-counts> <alpha> <betas>
For example:
$ACEPLUS/logistic-regression.R hex-counts.txt 0.5 betas.txt
NOTE: if this script fails to execute, you might not have Rscript installed, or your R environment might not be properly set up. In that case, you can run this script via the command "R CMD $ACEPLUS/logistic-regression.R hex-counts.txt 0.5 betas.txt". You may need to specify a full path to the R executable on your system.
Alpha specifies the type of regularization, just as with the logreg-splice.py command earlier; we recommend a default value of 0.5 to equally interpolate between ridge regression (0.0) and lasso regression (1.0). This script may run for many hours, depending on the number of training examples given. When running on 10,000 exons and 10,000 introns, we have found that the script takes approximately 12 hours on a single CPU core. When the script finishes, the learned hexamer weights should be in the betas.txt file:
4097 x 1 sparse Matrix of class "dgCMatrix"
1
(Intercept) 3.41155949
AAAAAA -47.12145843
AAAAAC .
AAAAAG 42.45011428
AAAAAT .
AAAACA -3.10915854
AAAACC .
AAAACG 0.86292402
...etc...
Now these weights need to be installed into model files for exons and introns. Because the intron weights are simply the negatives of the exon weights, the same betas.txt file generated above can be used for both exons and introns:
$ACEPLUS/install-logistic-features-in-sensor.py
betas.txt EXON exon.model
$ACEPLUS/install-logistic-features-in-sensor.py betas.txt INTRON intron.model
$ACEPLUS/install-logistic-features-in-sensor.py betas.txt INTRON intron.model
After verifying that the generated model files are non-empty, please copy them into the directory where your configuration script is located (see the next section).
Editing the Configuration File
A default configuration file called aceplus.config is included in the GitHub distribution. Please start with this default file and edit it as necessary. Below we describe the most important settings. Please put this file in a separate directory where your model files are located.For parameters that name a file, please give full absolute paths (unless you are using Docker, in which case you should omit paths, place all files in the current directory, and specify the vcf directory as /root).
min-path-score : Any gene prediction that scores less than this value will be omitted from the output of SGRF. The lowest possible score is 0.0, which is the default threshold. The highest possible value is 1.0.
max-alt-structures : This is the number of alternate splicing patterns that can be predicted by SGRF.
exon-model : this is the exon.model file generated above.
intron-model : this is the intron.model file generated above.
donor-model : this is the donors.model file generated above.
acceptor-model : this is the acceptors.model file generated above.
allow-exon-skipping : set this to true to allow prediction of exon skipping when a genetic variant disrupts an annotated splice site.
allow-cryptic-sites : set this to true to allow prediction of cryptic splice sites when a genetic variant disrupts an annotated splice site.
allow-denovo-sites : set this to true to allow prediction of novel splice sites present in the alternate sequence but not present in the reference.
allow-intron-retention : set this to true to allow prediction of intron retention when a genetic variant disrupts an annotated splice site.
donor-consensus : set this to the list of allowable donor splice site consensuses. For example: GT,AT,GC
acceptor-consensus : set this to the list of allowable acceptor splice site consensuses. For example: AG,AC
max-splice-shift : this is the maximum distance between a cryptic splice site and an annotated splice site. A recommended default is 150, as the majority of documented cryptic sites are within +/- 150bp of the annotated splice site.
intron-model : this is the intron.model file generated above.
donor-model : this is the donors.model file generated above.
acceptor-model : this is the acceptors.model file generated above.
allow-exon-skipping : set this to true to allow prediction of exon skipping when a genetic variant disrupts an annotated splice site.
allow-cryptic-sites : set this to true to allow prediction of cryptic splice sites when a genetic variant disrupts an annotated splice site.
allow-denovo-sites : set this to true to allow prediction of novel splice sites present in the alternate sequence but not present in the reference.
allow-intron-retention : set this to true to allow prediction of intron retention when a genetic variant disrupts an annotated splice site.
donor-consensus : set this to the list of allowable donor splice site consensuses. For example: GT,AT,GC
acceptor-consensus : set this to the list of allowable acceptor splice site consensuses. For example: AG,AC
max-splice-shift : this is the maximum distance between a cryptic splice site and an annotated splice site. A recommended default is 150, as the majority of documented cryptic sites are within +/- 150bp of the annotated splice site.
The remaining parameters are described on the ACE web page. Note that human model files for SGRF are included in the git distribution for ACE. If your target organism is human, you can use these files:
exons.model : logistic regression exon model
introns.model : logistic regression intron model
logreg-donors.model : logistic regression donor site model
logreg-acceptors.model : logistic regression acceptor site model
intergenic.model : intergenic model
If your target organism is not human, please retrain SGRF by following the instructions above on training the splice site models and the exon definition model. Note that the intergenic model need not be retrained, since SGRF cannot predict changes to the intergenic region.
Running SGRF
Once the models have been trained and the configuration file has been edited, you can run SGRF using this command:$ACEPLUS/aceplus.pl
config.cfg ref.fasta alt.fasta annotations.gff
max-VCF-errors outfile
The usage of this command is nearly identical to the usage of the original ACE command. Please see the original ACE web page for instructions on setting these parameters and interpreting the outputs.
Before running SGRF, you will need to make the personal genome (alt.fasta) using the make-individual-genomes.pl script, as described on the original ACE web page.
Getting Help
If you encounter difficulties, please email bmajoros@duke.edu.